Methods
FLAPW

An accurate and realistic description of materials of scientific or technological interest requires ab-initio methods that are able to handle a variety of phenomena such as non-collinear magnetism, spin-orbit coupling effects, (external) electric fields, correlation effects, low dimensions, etc. Within the vector spin-density formulation of density functional theory we developed a program, FLEUR, which allows us to investigate materials properties on a quantum mechanical level. This massively parallelized program is based on the full-potential linearized augmented planewave (FLAPW) method for bulk, film and wire geometry. With this method, it is possible to accurately describe a wide variety of systems with open structures and low symmetry. Force calculations enable us to simultaneously determine the magnetic and structural ground state.
Orbital-Dependent Density Functionals
For many years, local and semilocal density functionals, such as the local-density approximation and the generalized gradient approximation, have been the standard in electronic structure calculations based on density functional theory. With the advent of increasingly powerful computers, more sophisticated nonlocal orbital-dependent functionals are becoming more and more popular. Their simplest variants are the hybrid functionals, which contain a certain fraction of exact exchange admixed with local or semilocal functionals. The self-interaction error is thus partially canceled, which improves the description of strongly correlated materials and oxides. We have implemented two of the most popular hybrid functionals (PBE0 and HSE) into the FLEUR code. A treatment of orbital-dependent functionals (e.g., the exact exchange functional) within the Kohn-Sham formalism, which requires the effective potential to be purely local, is enabled by the optimized-effective-potential (OEP) method. A novel incomplete-basis-set correction has made the calculations particularly efficient and stable.The next logical step would be to add an orbital-dependent correlation functional, whose nonlocality and frequency dependence will make it possible to account for the van-der-Waals interaction including the dispersive force created by fluctuating dipoles. With the adiabatic-connection fluctuation-dissipation theorem we can make a connection to many-body perturbation theory as it allows to construct density functionals in a systematic manner from the frequency-dependent density-density correlation function, which can be expanded in terms of Feynman diagrams.
(M. Betzinger, M.Schlipf, C. Friedrich )
Exact Diagonalization
A straightforward approach to solving the many-body problem is to simply diagonalize the Hamiltonian. Of course this can only be done for finite systems, as the Hamiltonian is a matrix of finite dimension. Though finite, this dimension grows with increasing system size, to astronomical proportions. Already for fairly small clusters, tens of gigabytes of memory are needed for storing even a single many-body wave-function. Thus, while providing an exact solution to the many-body problem, it is very difficult to eradicate finite-size effects without using extremely large computers. In our calculations we use the Lanczos method to calculate the ground state, density matrix, spectral function, and dynamical responses.
(E. Koch)
Quantum Monte Carlo

For large systems, the Hilbert space becomes prohibitively large; it is then no longer possible, for example, to calculate the exact product of the Hamiltonian matrix with a state vector. The basic idea of the quantum Monte Carlo approach is to evaluate such matrix-vector products in a stochastic way. If all matrix elements of the Hamiltonian are positive, the ground state of very large systems can be determined exactly, within controllable statistical errors. For electrons, however, there are also always negative matrix elements in the Hamiltonian. In the quantum Monte Carlo approach, these give rise to the infamous sign-problem, which, if untreated, makes calculations for fermions impossible. To avoid the sign-problem, we use the fixed-node approximation. We have applied quantum Monte Carlo for calculating the ground state, static response functions, and quasiparticle energies.
(E. Koch)
The KKR Methode

The KKR method of band structure calculations was originally introduced in 1947 by Korringa and in 1954 by Kohn and Rostoker. A characteristic feature of this method is the use of multiple scattering theory for solving the Schrödinger equation. In this way, the problem is split into two parts. First, one solves the scattering problem of a single potential in free space. Second, one solves the multiple scattering problem by demanding that the incident wave to each scattering centre should be the sum of the outgoing waves from all other scattering centres.
The scheme has met with great success as a Green function method, within density-functional theory. Its applications range from the full potential ab-initio treatment of bulk, surfaces, interfaces and layered systems with O(N) scaling to the embedding of impurities and clusters in bulk and on surfaces. The method has been used with considerable success in the study of non-collinear magnetic structures, lattice relaxations, relativistic effects, and transport properties of solids.
“Minimum Search Nudged elastic band” Optimiser (MsNEB)
Optimisation is essential in many scientific and economic fields. However, optimisation problems are usually too complex to be solved using direct calculations or with trial and error. Two well-known methods that can find deep minima in complex systems are the simulated annealing method and the genetic algorithm.
In these methods, artifical fluctuations control the probability of escaping a local minimum of a certain depth. Here, a complementary method is developed based on the "Nudged Elastic Band" method, which is commonly used to determine saddle points. Here, the probability of escaping a local minimum depends on stretch rather than depth.
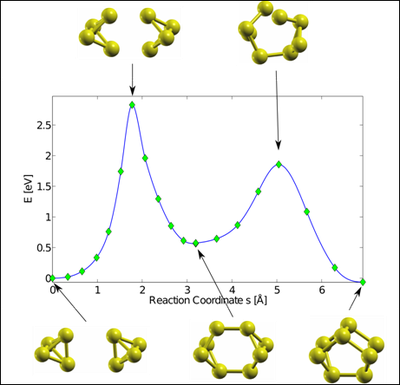
This recent method has already demonstrated its capability by finding, within the framework of density funtional theory (DFT), the most stable isomers of phosphorus P4, P8 molecules, as well as the corresponding molecules of Asn, Sbn, und Bin (n = 4,8). In the case of n = 8 , the method found stable and metastable configurations, some of which were new and have similar energy. As a by-product, an upper limit for the energy barrier between these configurations is obtained.
( J.Hischfeld, H.Lustfeld)